
En el primer año de vida, y especialmente en los primeros episodios, es frecuente confundir el síndrome de Dravet con crisis epilépticas febriles. La diferencia entre una y otra entidad es relevante, ya que en el caso de las crisis epilépticas febriles no se trata de una encefalopatía epiléptica del desarrollo, por lo que no está indicado un tratamiento crónico con fármacos antiepilépticos. El síndrome de Dravet, en cambio, precisa de medidas de prevención de la fiebre, cobertura antiepiléptica y tratamiento agresivo inicial ante la posibilidad de un estado epiléptico, así como la puesta en marcha de un programa de atención temprana, por lo que es importante que el paciente sea tratado adecuadamente y por un especialista con la menor demora posible.
Hasta hace relativamente poco, el diagnóstico solo era posible hacia los 2-4 años de vida debido a la necesidad de esperar a la evolución para poder determinarlo, pero el desarrollo del test genético, el estudio molecular y el mejor conocimiento de la clínica pueden adelantarlo actualmente.
Aunque una primera crisis con fiebre en el síndrome de Dravet puede ser similar a una crisis epilépticas febril, hay, no obstante, algunas particularidades en el síndrome de Dravet que permiten sospecharlo. Las crisis febriles del síndrome de Dravet aparecen en muchos casos antes de los 7 meses, tienden a ser prolongadas y a repetirse en un período breve. Además, suelen ser hemiclónicas, aunque las clónicas bilaterales o generalizadas tampoco son infrecuentes. La temperatura que las desencadena puede no ser excesivamente elevada.
Aproximadamente, a un 20% de los pacientes con síndrome de Dravet no se le encontrará mutación genética en el gen SCN1A.
Aspectos a tener en cuenta en el diagnóstico del síndrome de Dravet:
- Edad de inicio de las crisis epilépticas
- Las crisis epilépticas se desencadenaron por fiebre, enfermedad, calor o emoción a la actividad
- Tipos de crisis
- Frecuencia de las crisis
- Duración de las crisis
- Las crisis no han sido controladas por la medicación
- Algunos de los medicamentos empleados para las crisis epilépticas empeoraron el cuadro
- Aparición de otro tipo de crisis diferentes a las iniciales
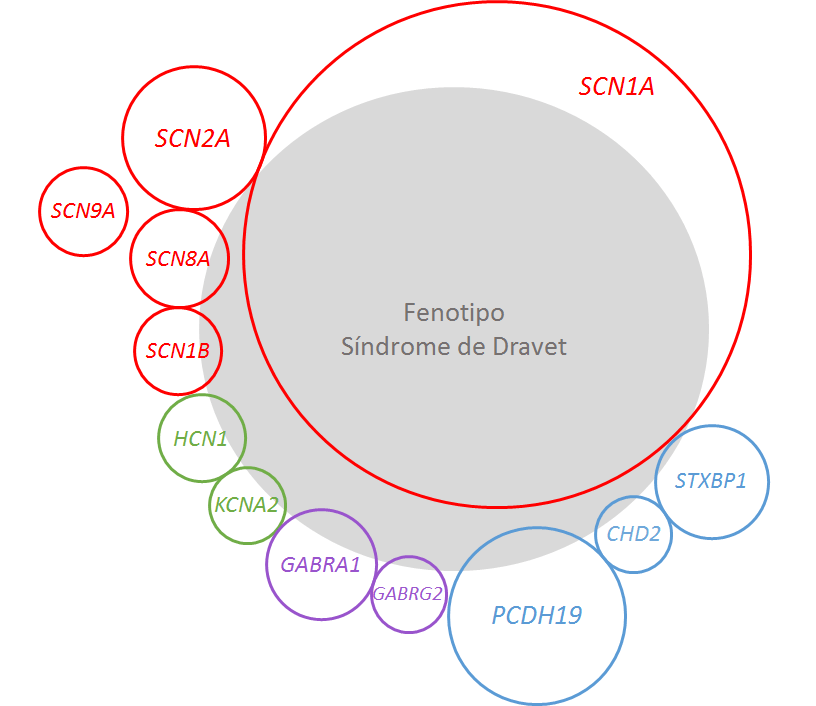
Alrededor del 80% de los pacientes diagnosticados con síndrome de Dravet tienen una mutación en el gen SCN1A. Este gen codifica la producción de canales de iones de sodio, que son proteínas de poros incrustadas en la membrana celular que permiten que los iones de sodio entren y salgan de la célula, propagando señales eléctricas. Algunos pacientes que parecen tener síndrome de Dravet tienen otras mutaciones, incluyendo SCN2A y PCDH19, que se corresponden con otros síndromes específicos.
- El 90% de las mutaciones del SCN1A son de novo, lo que significa que no se ha heredado y no se encuentra en los padres del paciente.
- Del 4 al 10% de las mutaciones del SCN1A se heredan de los padres. En este caso, hay un 50% de probabilidad de pasar la mutación a futuros niños.
- Hay más de 6.000 posiciones para que ocurra una mutación en el gen SCN1A. Por lo tanto, la mayoría de las mutaciones de los pacientes no se han reportado en otras personas.
- Se puede observar cualquier tipo de mutación SCN1A en el síndrome de Dravet, y el tipo de mutación no predice la gravedad de la enfermedad.
- Las mutaciones en el gen SCN1A también se asocian con migrañas, crisis epilépticas febriles (FS) o epilepsia generalizada con crisis epilépticas febriles plus (GEFS+, por sus siglas en inglés).
El síndrome de Dravet es un trastorno del neurodesarrollo que se caracteriza por una epilepsia severa resistente al tratamiento que presenta las siguientes características clínicas y electroencefalográficas:
– Inicio en el primer año de vida
– Desarrollo cognitivo normal previo al inicio de la crisis
– Resistencia al tratamiento farmacológico
– Crisis epilépticas prolongadas (más de diez minutos)
– Normalidad inicial con deterioro posterior del EEG, asociado a deterioro cognitivo progresivo con ataxia y otras alteraciones motoras
En la mayoría de los casos, las crisis epilépticas comienzan en el primer año de vida. Las primeras crisis están relacionadas con la aparición de fiebre y son convulsiones generalizadas tónicas-clónicas o hemiclónicas. En muchas ocasiones estas crisis desembocan en estatus epilepticus (episodios de larga duración o con repetición sin recuperación de la conciencia). Con el tiempo, también aparecen crisis afebriles o relacionadas con otros estímulos, y otro tipo de crisis epilépticas como las mioclonías, ausencias atípicas y crisis parciales-complejas.
A partir del segundo año se empiezan a observar síntomas de retraso en el desarrollo cognitivo y psicomotor. En muchos casos se observan ataxia, trastornos incluidos dentro del espectro autista, problemas alimenticios, de crecimiento y trastornos del sueño. El habla suele ser una de las facultades más afectadas.
Existe un alto porcentaje de casos que no cumplen todos los requisitos señalados. También se hallan otras formas de epilepsia de inicio clínico similar pero que no evolucionan de forma tan negativa.
Factores desencadenantes de las crisis en el síndrome de Dravet
– Cuadro de fiebre
– Cambios bruscos de la temperatura corporal (baño, calor, ejercicio físico…)
– Determinados estímulos como patrones visuales, luces, etc.
– Emociones intensas
¿Qué son las crisis epilépticas?
Las crisis o ataques epilépticos son episodios de alteración de la función del cerebro en forma de hiperexcitabilidad neuronal. Durante ellas, la señal eléctrica que utilizan las neuronas para comunicarse entre sí se propaga de forma desordenada y excesiva a otras neuronas vecinas e incluso a regiones alejadas. Las crisis epilépticas afectan a la corteza cerebral, donde se albergan funciones cerebrales que requieren de nuestra voluntad (por ejemplo, hablar, entender, pensar, memorizar, mover los músculos, prestar atención) y es la que recibe información de los órganos de la visión, el oído, el tacto, el olfato y el gusto. Cuando se produce una crisis epiléptica se alteran una o varias de estas funciones, por este motivo, los síntomas durante las crisis epilépticas son muy variables y reflejan la función de la región de corteza cerebral que ha provocado el inicio de la crisis y las funciones de las zonas donde se propaga.
Las crisis suelen tener una duración breve, menos de dos minutos, a veces sólo unos segundos. Inmediatamente después de una crisis las neuronas afectadas pierden transitoriamente su función, pues durante la excitación eléctrica consumen toda su energía. Por este motivo, después de cada crisis los enfermos suelen encontrarse cansados, somnolientos, confusos, con debilidad en alguna parte del cuerpo o con dificultad para hablar durante minutos o varias horas. Pasado este tiempo las neuronas recuperan la energía y reanudan su función normal.
¿Qué es un estatus epilepticus?
El estatus epilepticus se ha definido tradicionalmente como crisis que persisten por más de 30 minutos o 2 o más crisis repetidas sin recuperación de la conciencia entre ellas. Es una emergencia neurológica que requiere una atención inmediata. El diagnóstico y el tratamiento deben ser continuos a lo largo de los primeros minutos hasta su resolución. El estatus epiléptico puede ser convulsivo o no convulsivo y la monitorización electroencefalográfica continua es de gran ayuda para el diagnóstico y para valorar la respuesta al tratamiento.
Tipos de crisis
Las crisis epilépticas se dividen en tres grupos según su inicio: inicio focal, inicio generalizado e inicio desconocido. Las crisis focales se originan en una región circunscrita de la corteza cerebral, y desde ahí pueden propagarse a otras regiones. Si se asocian con confusión o alteración de la conciencia se denominan crisis parciales complejas, si no se asocian con alteración de la conciencia, son crisis parciales simples. Las crisis generalizadas se originan simultáneamente en los dos hemisferios cerebrales y se asocian con alteración de la conciencia.
Los tipos de crisis epilépticas más habituales en el síndrome de Dravet son:
– Crisis tónico-clónicas. Son crisis generalizadas (implican los dos hemisferios cerebrales). Comienzan con una rigidez en brazos y piernas (fase tónica), seguido de movimientos espasmódicos en brazos, piernas y cabeza (fase clónica). Los afectados pierden la consciencia durante la crisis epiléptica. Estos episodios suelen ser habituales en el síndrome de Dravet a cualquier edad.
– Mioclonías. Las mioclonías son movimientos involuntarios, breves, parecidos a sacudidas que provocan una contracción muscular brusca. Son consideradas crisis generalizadas, aunque en alguna ocasión pueden ser focales, causando movimientos bruscos de cabeza, brazo, o párpados.
– Crisis focales. Las crisis focales afectan a un área limitada del cerebro y el afectado mantiene la consciencia. En el caso de una crisis parcial compleja se pierde dicha consciencia.
– Crisis de ausencia. La mayoría de las ausencias típicas duran solo unos pocos segundos, suelen ser episodios de mirada fija o “ausencias”. Estos episodios pueden ocurrir muchas veces al día. Las ausencias atípicas comienzan de manera más lenta y duran más.
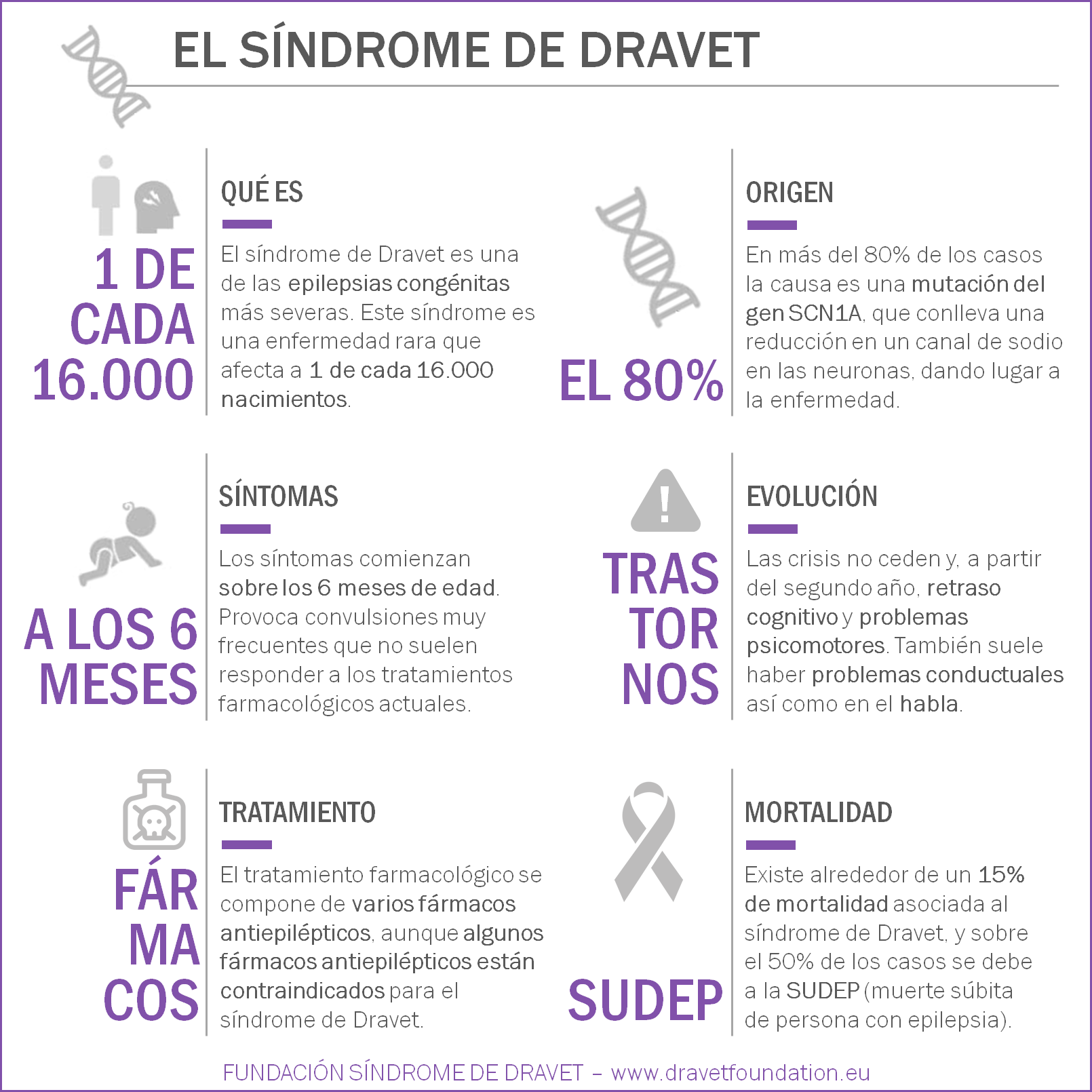
El síndrome de Dravet no se describió hasta finales de 1970 y hasta 2003 no existió un test genético que ayudara a diagnosticar la enfermedad. Esto explica que el número de afectados no se conozca con exactitud. Se estima que la incidencia de la enfermedad es de 1 de cada 16.000 nacimientos, lo que la encuadra en el grupo de enfermedades raras (afecta a menos de 1/2.500).
Aproximadamente un 25% de los pacientes presenta una historia familiar de epilepsia. Según los últimos estudios, se calcula que en España hay entre 348 y 540 pacientes correctamente diagnosticados. Sin embargo, los datos de prevalencia de la enfermedad nos invitan a pensar que el número de pacientes diagnosticados debiera ser superior a 2.000.
La evolución de la enfermedad es diferente en cada individuo. El síndrome de Dravet es un tipo raro de epilepsia de por vida. Los primeros signos de este síndrome son crisis epilépticas durante el primer año de vida en un niño con un desarrollo normal. Las crisis epilépticas varían, pero generalmente persisten durante toda la vida del paciente. Además, este síndrome afecta el desarrollo psicomotor y a las funciones cognitivas. Las funciones motriz, cognitiva y de comunicación se estabilizan pero los retrasos significativos permanecen en diversos grados.
Curso clínico
El síndrome de Dravet suele tener una evolución de crisis epilépticas dependiente de la edad. Se pueden identificar tres etapas:
– La fase de inicio (niños menores de un año de edad)
– La fase de empeoramiento (niños entre 1 y 5 años)
– La fase de estabilización (niños mayores de cinco años)
1/ FASE DE INICIO (niños menores de 1 años de edad)
El primer evento epiléptico generalmente ocurre «de la nada» normalmente entre los 4 y 8 meses de edad en un niño que se desarrolla con normalidad y no tiene antecedentes que sugieran un problema. En la mayoría de los casos, la primera crisis es inducida por una fiebre leve (37-38 ° C) a causa de una enfermedad o después de una vacunación. Por lo general, se trata de una crisis epilépticas clónica, ya sea inicialmente generalizada o solo en un lado del cuerpo (hemiclónica). Si es de un lado, puede permanecer en ese lado o extenderse a ambos lados y generalizarse. La primera convulsión suele ser una convulsión prolongada (> 15 minutos) que a veces se convierte en un estado epiléptico (> 30 minutos).
Durante las próximas semanas o meses, el niño tendrá otras crisis epilépticas, febriles o afebriles, a pesar del uso de medicamentos antiepilépticos. En este punto, el electroencefalograma (EEG) y otras investigaciones de neuroimagen (resonancia magnética) casi siempre son normales.
2/ FASE DE EMPEORAMIENTO (niños entre 1 a 5 años de edad)
Durante este período, la frecuencia de las crisis epilépticas suele aumentar. Las crisis epilépticas pueden ocurrir con o sin fiebre, y con frecuencia incluyen episodios de estado epiléptico (> 30 minutos).
Aparecen otros tipos de crisis epilépticas: crisis epilépticas mioclónicas, ausencias atípicas y convulsiones focales. Algunos factores ambientales pueden desencadenarlos: destellos de luz excesivos o intermitentes o patrones y diseños especiales, como patrones geométricos regulares, líneas o puntos, esfuerzo físico o incluso excitación.
Durante esta fase, las crisis epilépticas suelen ser extremadamente frecuentes, intensas y prolongadas, lo que suele dar como resultado múltiples hospitalizaciones.
3. FASE DE ESTABILIZACIÓN (niños mayores de 5 años de edad)
Desde la mitad de la infancia hasta la adolescencia, las crisis epilépticas generalmente mejoran, con una reducción y, a veces, la desaparición de las crisis focales, las ausencias atípicas y las crisis mioclónicas. Las crisis epilépticas febriles convulsivas generalmente persisten, aunque el número de fiebres es menor.
El número y la duración de las crisis epilépticas disminuyen, pero las crisis epilépticas rara vez se detienen por completo. Estas convulsiones a menudo se agrupan, especialmente al principio o al final de la noche.
En la mayoría de los pacientes, el estado epiléptico es considerablemente menos frecuente.
Desarrollo psicológico y cognitivo
El síndrome de Dravet tiene una evolución del desarrollo característica. El retraso en el desarrollo y la discapacidad intelectual casi siempre están presentes.
Al igual que en el curso clínico de las crisis epilépticas, se pueden identificar tres etapas, aunque las edades pueden variar:
– La fase de inicio (niños menores de un año de edad)
– La fase de empeoramiento (niños entre 1 y 5 años)
– La fase de estabilización (niños mayores de cinco años)
1/ FASE DE INICIO (niños menores de 1 años de edad)
El desarrollo psicomotor y cognitivo del niño suele ser normal durante el primer año de vida.
2/ FASE DE EMPEORAMIENTO (niños entre 1 a 5 años de edad)
Entre el primer y segundo año de edad, a menudo se observa una desaceleración general del desarrollo. Aparecen trastornos intelectuales y de comportamiento. Su severidad puede variar de un niño a otro. El habla y el lenguaje son los primeros en verse afectados, pero la mayoría de las otras áreas de desarrollo se ven afectadas gradualmente.
El niño está a menudo de mal humor, hiperactivo, contrario, terco y obstinado. Los problemas de comunicación dificultan aún más la interacción social. Los trastornos del sueño son comunes.
Muchos de los niños tienen problemas con la coordinación, como la marcha descoordinada (ataxia) y problemas con las habilidades motoras finas (coordinación de las manos y los dedos).
3/ FASE DE ESTABILIZACIÓN (niños mayores de 5 años de edad)
Al llegar a esta fase, los trastornos psicomotores y cognitivos tienden a estabilizarse. El desarrollo puede continuar lentamente o reiniciarse si ha habido un período de regresión.
La discapacidad intelectual permanente varía de moderada a severa, dependiendo de la evolución durante los años anteriores.
La comunicación a menudo sigue siendo difícil y se pueden observar ciertas características del espectro autista. En general, las habilidades del habla coinciden con el nivel intelectual general del niño, pero la comprensión del lenguaje sigue siendo mejor que la capacidad para producir el habla.
Ocasionalmente pueden tener un comportamiento agresivo o incluso psicosis.
Desarrollo a largo plazo
La historia del síndrome de Dravet se remonta a 1978, cuando fue inicialmente descrito por la epileptóloga Charlotte Dravet como Epilepsia Mioclónica Severa de la Infancia. No fue hasta 1991 que se reconoció como síndrome de Dravet y hasta 2001 no se confirmó que el síndrome de Dravet es de origen genético. Debido a ello, la evolución a largo plazo y la esperanza de vida del síndrome de Dravet no son bien conocidas.
Sin embargo, según la experiencia clínica, el manejo temprano y apropiado del síndrome de Dravet parece dar lugar a un mejor resultado.
La mortalidad asociada al síndrome de Dravet ha sido descrita en los últimos estudios con porcentajes alrededor del 15%, debido principalmente a la SUDEP (muerte súbita inesperada en la epilepsia), convulsiones prolongadas, accidentes relacionados con convulsiones como ahogamiento e infecciones. La SUDEP representa actualmente la mitad de los casos de mortalidad en el síndrome de Dravet.
En los últimos años, se ha producido una leve mejoría de los datos, que puede ser debida a un mejor manejo de las crisis y a una mejor elección de los fármacos antiepilépticos.
Si bien una mutación no es necesaria para el diagnóstico, puede respaldar un diagnóstico clínico y es útil para comprender el ADN, los genes y las mutaciones. Cada persona tiene dos copias del gen SCN1A: una de cada progenitor. Muchas mutaciones encontradas en el síndrome de Dravet hacen que una copia sea disfuncional, dejando solo una copia funcional. Esto da lugar a una condición llamada haploinsuficiencia, lo que significa que una copia en funcionamiento no es suficiente para prevenir los síntomas. Aproximadamente, el 90% de las mutaciones de Dravet son de novo, lo que significa que no se heredan del progenitor, sino que son nuevas mutaciones en el niño.
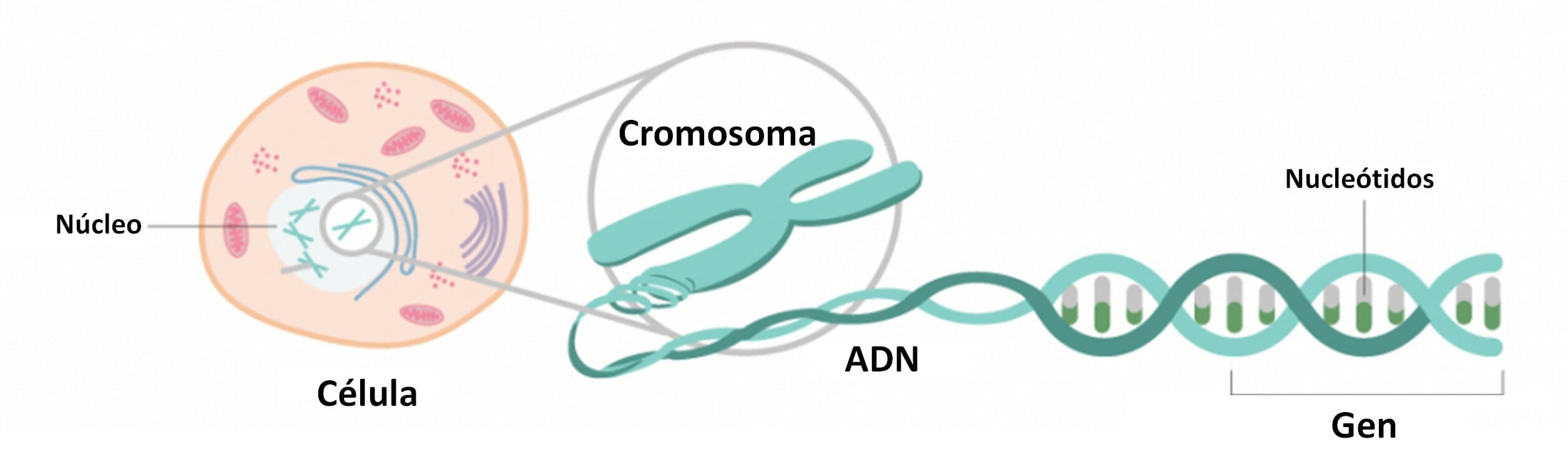
¿Qué es el ADN?
El ADN es el conjunto de instrucciones contenidas dentro de cada una de las células de nuestro cuerpo. Las instrucciones le dicen a la célula cómo construir las proteínas que necesita para funcionar. Una cadena de ADN es una cadena larga de 4 nucleótidos diferentes (abreviada A, T, C y G) unidas en un orden particular, de miles de millones de nucleótidos. Debido a que hay mucho ADN en nuestras células, está organizado en 23 pares de cromosomas, al igual que dos conjuntos de enciclopedias se organizarían en 23 volúmenes cada uno. Cuando se combinan un espermatozoide y un óvulo, cada uno con 23 cromosomas, el resultado es un total de 46 cromosomas, organizados en 23 pares iguales.
¿Qué es un gen?
Los 23 pares de cromosomas se dividen en segmentos más pequeños llamados genes. Un gen es muy parecido a un capítulo en una enciclopedia y contiene las instrucciones para producir una proteína específica. Cada gen es un pequeño segmento de ADN y, por lo tanto, también es una cadena larga de 4 nucleótidos diferentes unidos en un orden particular. Debido a que nuestras células tienen una copia de cada gen de cada progenitor, cada célula tiene dos copias de cada gen a menos que el gen se transmita en el cromosoma X o Y que determina el sexo. El gen SCN1A no está en el cromosoma X o Y.
Los genes se leen en grupos de 3 nucleótidos llamados codones. Cada codón se traduce en uno de los veinte aminoácidos, que luego se unen como cuentas en un collar. Los aminoácidos interactúan entre sí según sus propiedades químicas similares a la forma en que las perlas magnéticas se atraen y se repelen entre sí cuando se pliegan en la mano. A medida que los aminoácidos interactúan, la cadena larga se pliega sobre sí misma para formar una forma tridimensional muy específica.
En el caso del síndrome de Dravet, el gen afectado es el SCN1A, cuya forma 3D es un canal iónico que funciona como un canal cerrado en la membrana celular, permitiendo que los iones de sodio entren y salgan de la célula. La entrada y salida de iones permite que las señales eléctricas se propaguen a lo largo de las neuronas. Las siglas, más concretamente, significan que este gen codifica la subunidad 1 alfa de los canales de sodio activados por voltaje presentes en las neuronas (Sodium ChaNnel protein type 1 subunit Alpha).
Es por esto que el canal recibe también el nombre de NaV1.1, donde “Na” es el símbolo químico del sodio y “V” hace referencia al hecho de que este canal se activa por voltaje; es decir, se abre cuando llega un potencial de acción, que es una señal eléctrica, y ayuda a que éste se propague a través de las neuronas, ya que es el encargado de transmitir la información entre éstas.
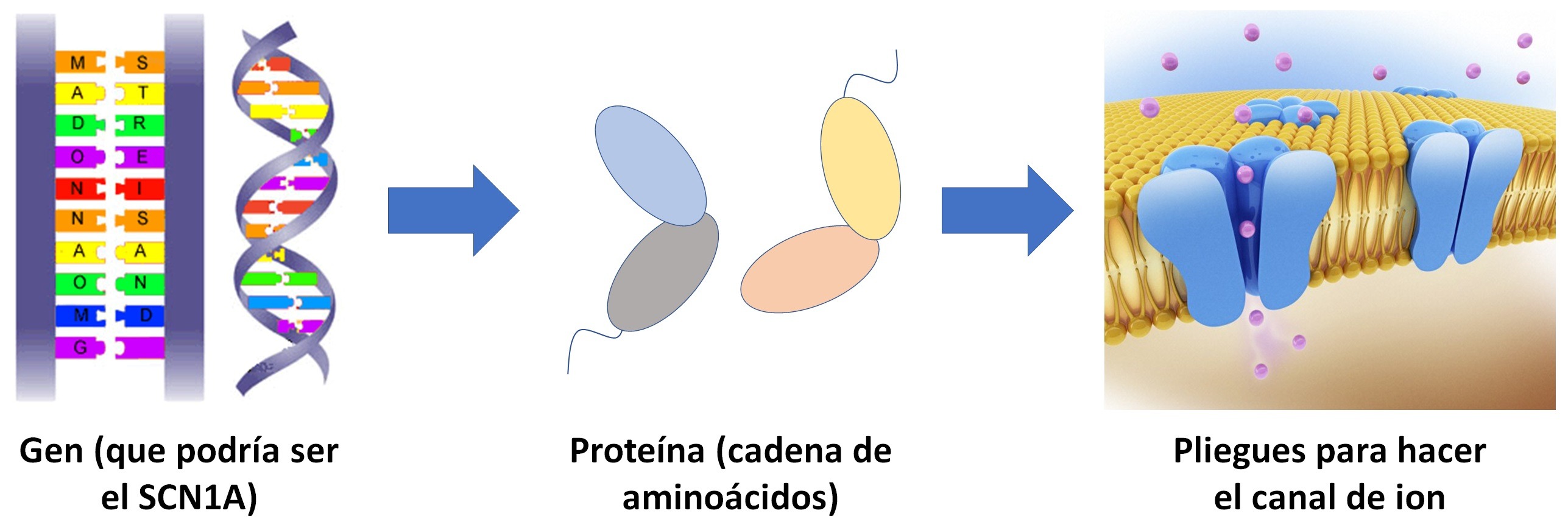
¿Qué es una mutación?
Una mutación es un cambio en la secuencia esperada de nucleótidos dentro de un gen. Este cambio en la secuencia original de ADN puede alterar la secuencia de aminoácidos, lo que puede hacer que la cadena termine prematuramente, se doble incorrectamente o altere de otro modo la funcionalidad del canal de sodio.
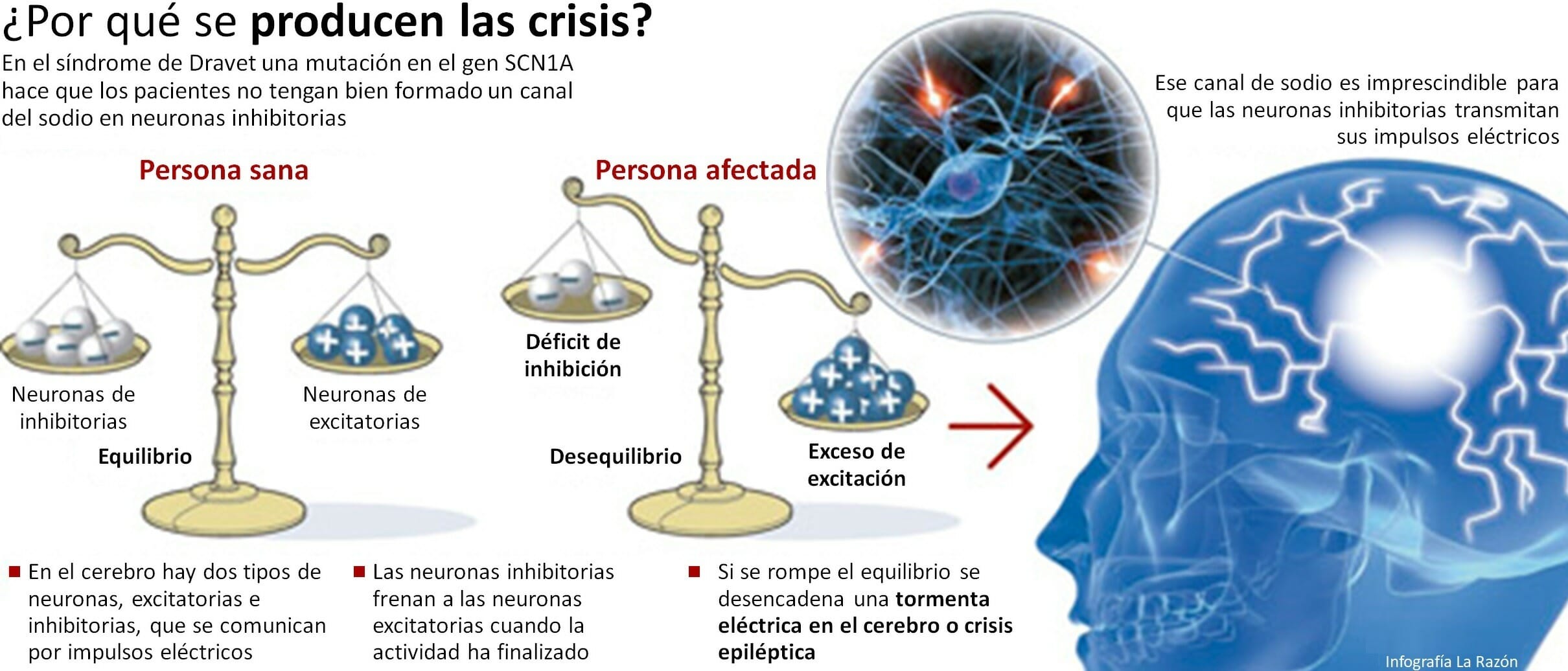
Como mencionábamos antes, la propagación del potencial de acción es esencial para el correcto funcionamiento del sistema nervioso y el cerebro los usa para procesar la información sensorial y controlar funciones corporales. Concretamente, en el SD, el canal afectado se encuentra en unas neuronas inhibitorias, conocidas como neuronas GABAérgicas por el neurotransmisor que liberan (GABA, por ácido gamma-aminobutírico). De esta forma, las alteraciones en SCN1A, que generan canales de sodio disfuncionales, provocan que estas neuronas no funcionen correctamente y, por lo tanto, se da una falta de inhibición de las neuronas excitatorias. Como resultado se obtienen una actividad eléctrica y crisis epilépticas inadecuadas, características del síndrome de Dravet.
Aunque el SCN1A tiene 160.000 nucleótidos, el cuerpo edita esta secuencia de 160.000 hasta aproximadamente 6.000 en la transcripción final de SCN1A, que sirve como las instrucciones para el canal de iones de sodio. Aún así, con más de 6,000 posiciones de nucleótidos, no es de extrañar que la mayoría de las mutaciones reportadas en la literatura científica aún no se hayan visto en otro paciente.
Recuerde que cada celda contiene dos copias de SCN1A; una de cada progenitor. Por lo general, solo se muta una copia, una condición denominada heterocigosidad. Aproximadamente del 4 al 10% de las mutaciones observadas en el síndrome de Dravet se heredan directamente de los padres, y los padres a menudo experimentan menos síntomas y menos graves que el niño en un fenómeno conocido como penetrancia reducida.
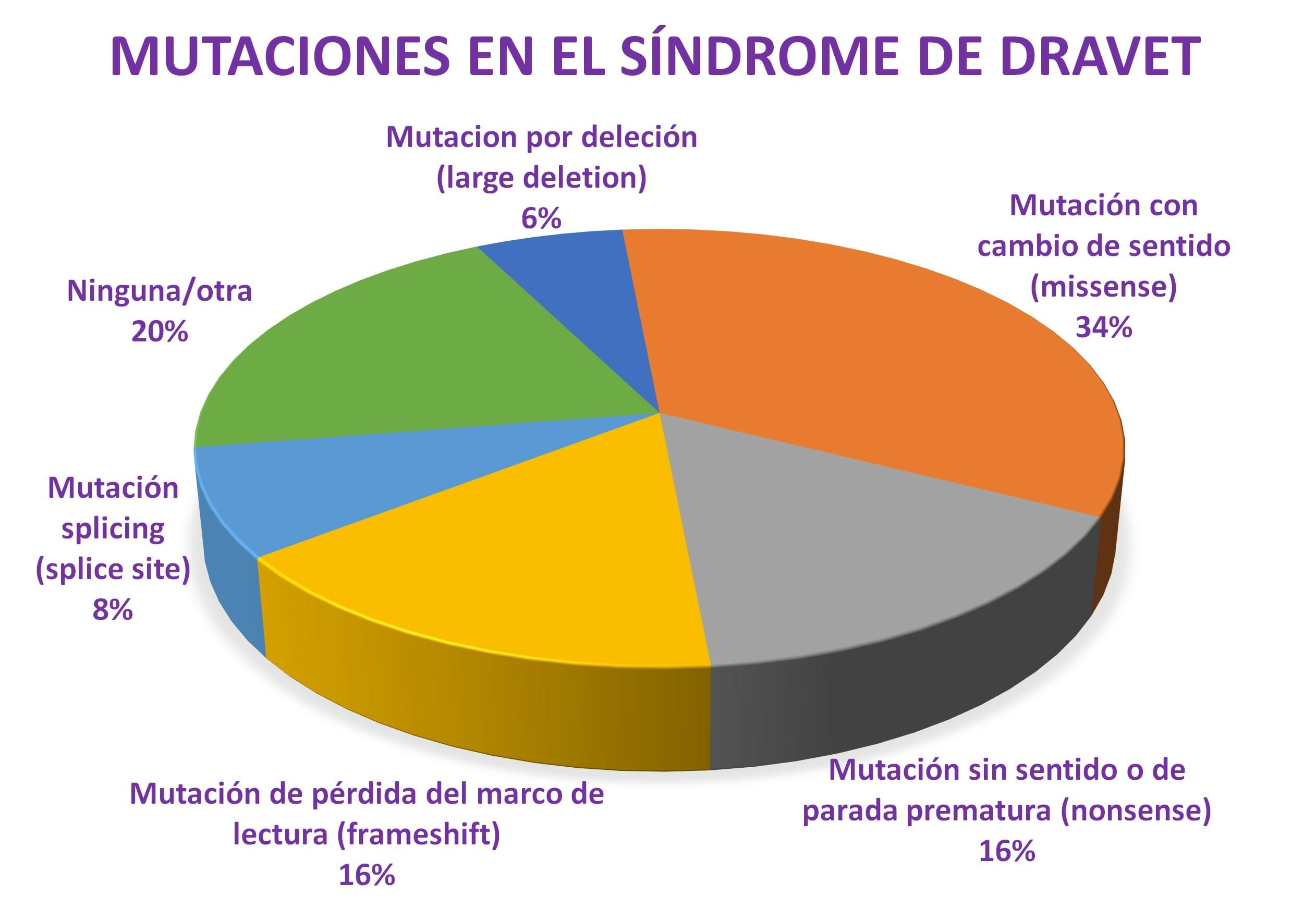
Hay tres tipos principales de mutaciones: missense, nonsense e inserciones/deleciones.
1.- Con cambio de sentido (missense)
Una mutación missense o con cambio de sentido es una simple sustitución de un nucleótido por otro en una única ubicación en un gen. Este ligero cambio en la secuencia de nucleótidos puede o no resultar en un aminoácido cambiado en la cadena larga.
Si se produce una mutación missense cerca de una región de formación de poros del canal de iones de sodio, es probable que altere significativamente la función del canal de iones y provoque un caso más grave de epilepsia relacionada con el SCN1A, como el síndrome de Dravet. Si se produce una mutación missense en una ubicación menos crítica del gen, puede producir síntomas clínicos más leves o no presentar ningún síntoma. Aproximadamente el 47% de las mutaciones observadas en pacientes con Dravet son mutaciones missense.
Una mutación sin sentido reportada por una compañía de pruebas genéticas puede verse así:
– Variante 1: Transversión G> T
– Posición del nucleótido: 4073
– Posición del codón: 1358
– Cambio de aminoácidos: triptófano > leucina
– Variante de significado desconocido (heterocigoto)
Esto dice que la mutación fue una sustitución de G por T en la posición del nucleótido 4073 (de 6000 en el gen final que se lee). Recuerde que los nucleótidos se leen en grupos de 3, llamados codones, por lo que 4073 dividido por 3 le da la posición del aminoácido o codón de 1358. El aminoácido leucina se sustituyó por el aminoácido triptófano. El laboratorio no puede determinar la importancia, ya que las mutaciones missense o con cambio de sentido se pueden asociar con presentaciones clínicas leves y graves. El paciente tiene una sola copia de esta mutación y, por lo tanto, es heterocigoto. Esta mutación de la vida real está, de hecho, en una región porosa del canal de ion de sodio, y este paciente tiene el síndrome de Dravet.
2.- Sin sentido o de parada prematura (nonsense)
Las mutaciones sin sentido o de parada prematura (nonsense) son similares a las mutaciones missense en que un nucleótido se sustituye por otro. Sin embargo, en el caso de una mutación nonsense, esa sustitución hace que el codón se lea como una señal de “PARADA”. La célula deja de leer el gen prematuramente y la proteína se acorta o trunca de manera significativa. Las mutaciones nonsense a menudo se asocian con epilepsias más graves relacionadas con el SCN1A, como el síndrome de Dravet. Aproximadamente el 20-40% de las mutaciones en el síndrome de Dravet son mutaciones nonsense (truncamiento). Una mutación nonsense puede ser reportada así:
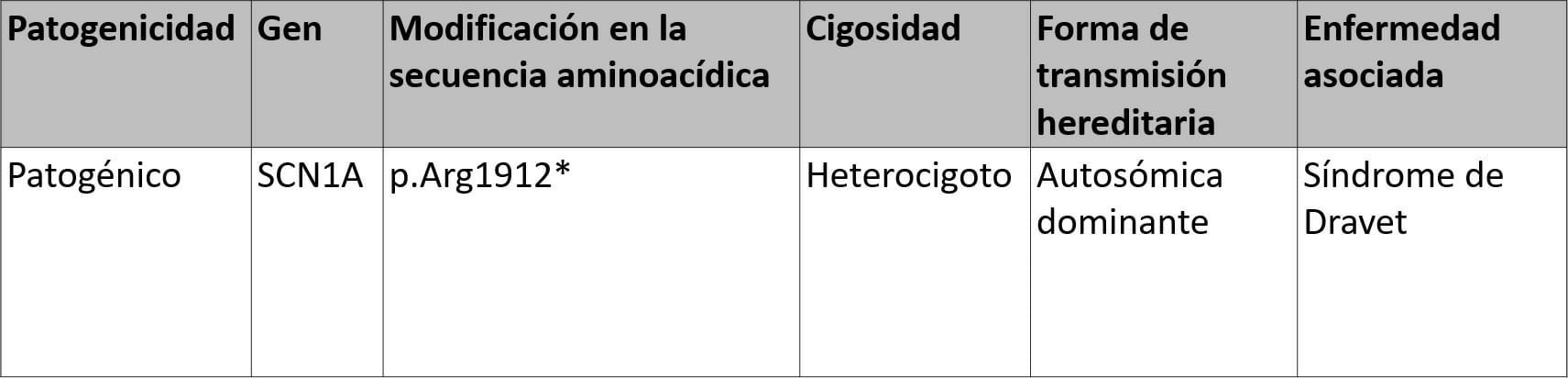
Este informe no especifica la posición del nucleótido, pero identifica la posición del aminoácido como 1912, y el asterisco (*) junto a la posición del aminoácido indica un codón de parada. En este caso, el laboratorio cree que esta mutación dará lugar al síndrome de Dravet.
3.- Inserción/deleción
A veces, uno o más nucleótidos se eliminan del gen. Si se insertan o eliminan uno o dos nucleótidos, el marco de lectura de los codones se desplaza, y cada aminoácido es incorrecto desde ese punto en la cadena. Esto usualmente causa una disfunción en el canal de sodio. Además, el cambio en el marco de lectura a menudo hará que uno de los codones más abajo de la cadena se interprete como un codón de parada, terminando prematuramente una cadena ya disfuncional.
Si se inserta o elimina un grupo de 3 nucleótidos, solo se agrega o elimina un codón, respectivamente, y la proteína aún puede ser funcional dependiendo de la ubicación de esa inserción o eliminación. Se pueden insertar o eliminar grandes segmentos de ADN, incluido el gen SCN1A completo y/o genes cercanos. Estas mutaciones tienen fenotipos variables, y representan aproximadamente el 2-5% de las mutaciones de Dravet.
Mosaicismo genético
Cuando la mutación se produce en el espermatozoide, en el óvulo o muy poco después de la fertilización, todas las células hijas derivadas del embrión en crecimiento contendrán la mutación. Este es el caso de la mayoría de las mutaciones encontradas en el síndrome de Dravet.
Sin embargo, si la mutación ocurre más tarde en el desarrollo del embrión, solo las células que descienden de la célula mutada llevarán la mutación. Las células que descienden de las células no mutadas del embrión permanecerán sanas. Esto se traduce en un individuo que es un mosaico de la mutación. Cuanto más tarde se produjo la mutación, menor es el porcentaje de células que descienden de la célula mutada y menor es el “% mosaicismo” o “carga de mosaico”. (Esta es una amplia generalización: en realidad, el grado de especialización de las células en el momento de la mutación juega un papel importante en el lugar donde las células mutadas se concentran en el cuerpo del paciente y cuál es la carga de mosaico final.) Un estudio informó que las mutaciones del gen SCN1A con una carga de mosaico del 12-25% eran potencialmente patógenas, con penetrancia reducida, lo que significa que no todos los que tenían la mutación en forma de mosaico exhibían signos o síntomas.
Polimorfismo de un solo nucleótido
Las mutaciones son en realidad un fenómeno natural que ha estado ocurriendo en todos los organismos durante miles de años. La mayoría de los cambios en la secuencia de ADN tienen poco o ningún efecto en los productos de la proteína final porque ocurren en regiones que se editan durante el procesamiento del gen, o su ubicación en la proteína final no altera su función. De hecho, muchos miembros de la población sana tienen variantes en sus genes que se comparten con un porcentaje significativo de la población. Debido a que estas variantes no tienen síntomas clínicos evidentes, se denominan polimorfismos de nucleótido único (SNP) y no se consideran mutaciones. Su informe de laboratorio puede incluir estos SNP, pero su presencia no se considera una prueba SCN1A positiva.
¿Qué significa esto para el paciente?
Los investigadores y los epileptólogos están aprendiendo más sobre el papel que desempeñan las mutaciones de SCN1A en el síndrome de Dravet y las epilepsias relacionadas todos los días. En este punto, está claro que las mutaciones de SCN1A de cualquier tipo pueden ser responsables del síndrome de Dravet. Sin embargo, debido a que algunas mutaciones de SCN1A están presentes en individuos con síntomas leves, probablemente hay muchos factores modificadores que determinan la gravedad de los síntomas que resultan de la mutación. Las mutaciones de SCN1A son útiles para respaldar un diagnóstico clínico, pero recuerde que aproximadamente el 20% de los pacientes con síndrome de Dravet no han detectado una mutación y no se requiere una mutación para el diagnóstico.
¿Son todo malas noticias?
¡En absoluto! Hay tanta investigación en marcha sobre el gen SCN1A, el síndrome de Dravet y epilepsias relacionadas que los científicos están descubriendo nuevos conocimientos y vías terapéuticas potenciales prácticamente todos los días. El hecho de que un síndrome de epilepsia como el Dravet pueda atribuirse a una causa raíz, a pesar de muchos factores y modificadores desconocidos, lo convierte en un objetivo atractivo para la investigación y brinda a los pacientes la esperanza de una cura cercana.
En la actualidad no existe cura para el síndrome de Dravet; sin embargo, es posible reducir el número y la gravedad de las convulsiones con tratamientos farmacológicos (medicamentos antiepilépticos) y evitando los factores desencadenantes. El tratamiento del síndrome de Dravet requiere un tratamiento integral. Además de los medicamentos, otros tratamientos no farmacológicos pueden ser útiles junto con medidas de atención integral. La familia juega un papel crítico.
Tratamientos
Todos los tratamientos deben realizarse bajo la supervisión de un médico con conocimientos sobre el síndrome de Dravet. Hay muchos tratamientos antiepilépticos en todo el mundo, pero es posible que algunos no lo estén o no estén aprobados oficialmente en nuestro país. Toda iniciación, cambio o modificación del tratamiento antiepiléptico debe realizarse solo con la aprobación del médico que sigue regularmente al paciente. Si tiene alguna pregunta sobre los tratamientos, no dude en consultar a este médico.
Se pueden usar varios tratamientos para pacientes con síndrome de Dravet: Farmacológicos y no Farmacológicos.
A/ TRATAMIENTOS FARMACOLÓGICOS
Los medicamentos antiepilépticos son un grupo diverso de productos farmacéuticos, utilizados en el tratamiento de las crisis epilépticas. En todo el mundo hay una gran cantidad de medicamentos antiepilépticos disponibles. Se dividen en varias clases. Su uso depende del trastorno epiléptico específico. Algunos pueden mejorar la situación del paciente, mientras que otros pueden empeorarlo. La disponibilidad de medicamentos específicos en España dependerá de las regulaciones gubernamentales y las decisiones de comercialización del fabricante farmacéutico. El síndrome de Dravet es una epilepsia altamente farmacorresistente, lo que significa que las convulsiones son difíciles de controlar, y que muchos pacientes se tratan con más de un medicamento antiepiléptico.
1/ Medicamentos frecuentes
Por experiencia, se conoce que el valproato y las benzodiacepinas son eficaces en la reducción de las crisis de estos pacientes. Se recomienda iniciar el tratamiento con valproato tras la primera crisis. Si no se consigue control de las crisis, se recomienda agregar topiramato o bien estiripentol. Tanto estiripentol como topiramato se suelen administrar añadiendo de base valproato y clobazam.
2/ Medicamentos a evitar
La lista a continuación indica algunos de los medicamentos que pueden empeorar las convulsiones y, por lo tanto, deben evitarse:
– Lamotriginia (Lamictal, Labileno, Crisomet)
– Fenitoína (Dilantin, Epanuntin, Sinergina)
– Lacosamida (Vimpat)
– Fosfenitoína (Cerevyx, Prodilantin)
– Carbamazepina (Tegretol, Calepsin, Cargagen, Barbatrol, Epitol, Finlepsin, Sirtal, Stazepine)
– Oxcarbazepina (Trileptal)
– Rufinamida (Inovelon)
– Tiagabina (Gabitril)
– Acetato de eslicarbazepina (Zebinix)
– Vigabatrina (Sabril, Sabrilan, Sabrilex)
3/ Tratamiento de emergencia
Las benzodiacepinas de emergencia se usan para evitar o detener las convulsiones de larga duración que pueden convertirse en estatus epilépticos.
Los tratamientos de emergencia en el hogar son en su mayoría fármacos antiepilépticos de acción rápida.
Su médico podrá recetarle un medicamento apropiado para detener las convulsiones de larga duración. Es necesario que un profesional experto en atención de la salud le indique con cuidado cómo usar este medicamento.
Existen diferentes formas de fármacos antiepilépticos de acción rápida: las vías de administración de estos medicamentos son nasales, orales o rectales.
Tratamiento de urgencias:
Si acude a la sala de emergencias, el médico puede usar otros tratamientos de acuerdo con el estado clínico, la práctica local y la disponibilidad de medicamentos de su hijo.
B/ OTRAS MEDIDAS ANTIEPILÉPTICAS
A veces, los medicamentos antiepilépticos no son suficientes para controlar las convulsiones. Después de hablar con su médico, se pueden considerar otros tratamientos además de los medicamentos:
– Dieta Cetogénica
– Estimulación del Nervio Vago
– Tratamiento con Inmunoglobulinas
Tenga en cuenta que cada niño con síndrome de Dravet es único y que lo que podría funcionar para un niño puede no funcionar para otro. Estos tratamientos no farmacológicos han mostrado resultados variables.
1/ Dieta cetogénica
La dieta cetogénica es una dieta rica en grasas, adecuada en proteínas y baja en azúcar. La dieta obliga al cuerpo a quemar grasas en lugar de azúcares. Normalmente, los azúcares contenidos en los alimentos se convierten en glucosa, que luego se transporta alrededor del cuerpo y es particularmente importante para alimentar la función cerebral.
Sin embargo, si hay muy poca azúcar en la dieta, el hígado convierte la grasa en ácidos grasos y cuerpos cetónicos. Los cuerpos cetónicos pasan al cerebro y reemplazan la glucosa como fuente de energía. Un nivel elevado de cuerpos cetónicos en la sangre, un estado conocido como «cetosis», puede llevar a una reducción en la frecuencia de las crisis epilépticas.
Existen varias variantes de esta dieta, que incluyen recetas caseras o fórmulas preparadas para consumir.
Es una dieta muy estricta con riesgos potencialmente significativos y que no debe hacer por su cuenta. Requiere el asesoramiento de un equipo con experiencia para maximizar los beneficios y minimizar los riesgos.
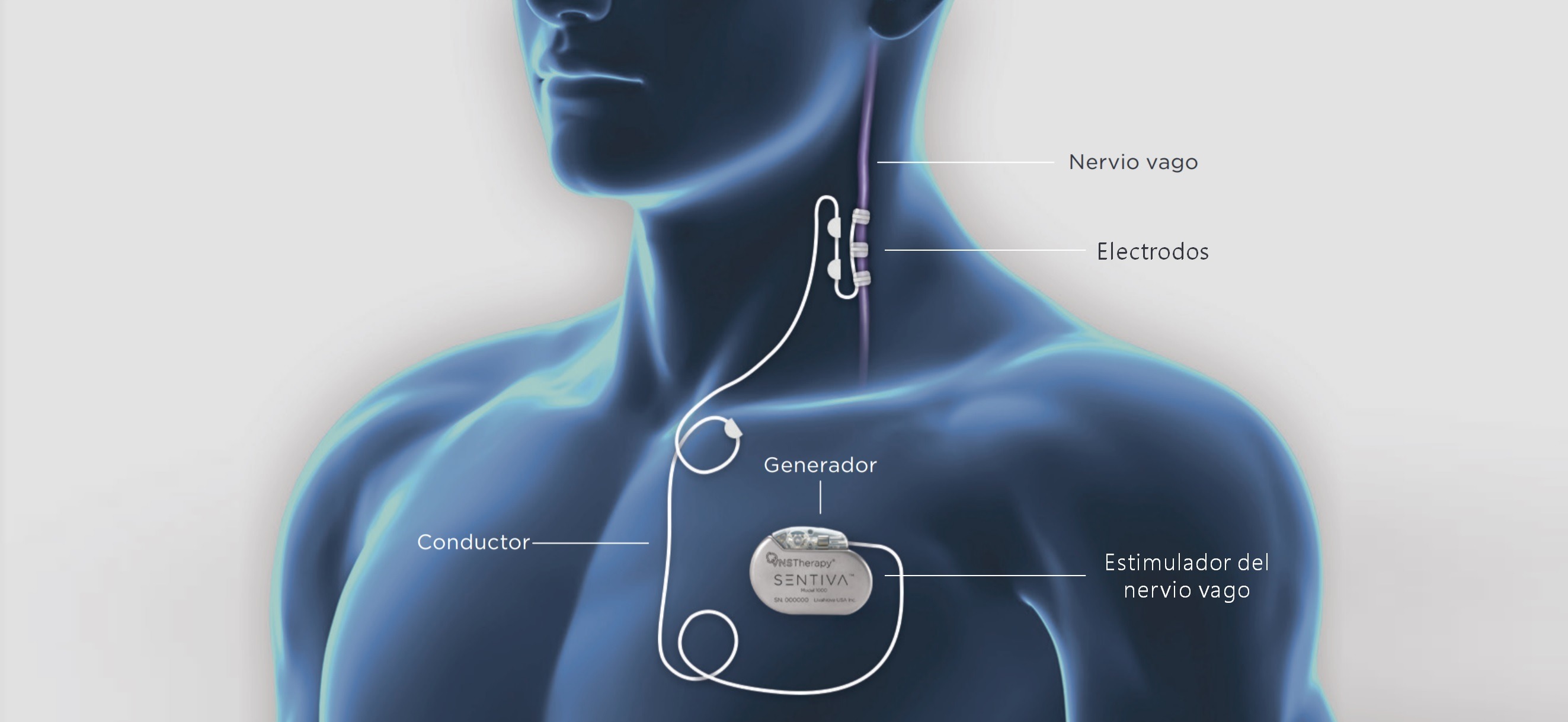
2/ Estimulación del nervio vago
El nervio vago es un nervio especial que transporta los impulsos motores del cerebro a varios órganos (pulmones, corazón, intestinos, vasos sanguíneos…) y viceversa. La estimulación vagal puede regular los ataques epilépticos por un mecanismo de acción desconocido.
La estimulación del nervio vago consiste en la implantación quirúrgica de un dispositivo debajo de la piel del tórax, similar a un marcapasos. Este dispositivo luego se conecta mediante un cable oculto debajo de la piel al nervio vago en el cuello.
3/ Tratamientos con inmunoglobulinas
El tratamiento con inmunoglobulinas intravenosas ha dado resultados dispares, logrando en alguna ocasión la supresión de las crisis o una disminución de estas. Normalmente este tratamiento se suele repetir cada 2-3 semanas durante 6 ocasiones. Los resultados suelen ser transitorios, lo que obliga a repetir el tratamiento.
Protocolo de emergencia
El síndrome de Dravet tiene un impacto importante en la vida familiar, especialmente cuando el niño tiene convulsiones prolongadas que requieren tratamiento hospitalario.
Cuando hay convulsiones graves de larga duración, es útil y necesario tener un plan bien elaborado para el niño con síndrome de Dravet, los padres y los hermanos.
Junto con expertos, desde la Fundación Síndrome de Dravet hemos desarrollado un protocolo médico de urgencia específico para el síndrome de Dravet, el cual recomendamos llevar siempre con el paciente para hacer uso de él en el caso de que tenga una crisis prolongada.
Este ejemplo de protocolo médico de urgencia para el síndrome de Dravet se puede descargar en el siguiente botón, para enviárselo al neurólogo, adaptarlo al paciente y llevarlo siempre junto con el paciente con síndrome de Dravet para entregárselo al personal de urgencias en el caso de que haya una crisis prolongada.
A/ PROTOCOLO EMERGENCIA PARA NIÑOS DRAVET
Es muy difícil predecir cuándo podría ocurrir un estatus epiléptico. Cuando salga de su hogar con su hijo Dravet, es recomendable que lleve al menos los siguientes artículos:
– Teléfono móvil para pedir ayuda
– Protocolo de emergencia para presentar a ambulancias y profesionales de salas de emergencia
– Tratamiento de rescate para detener las convulsiones prolongadas
– Pañales
– Pulverizador de agua para refrescar al niño si hace calor
– Barritas de cereal, bocadillos o snacks (las visitas a la sala de emergencia pueden ser largas y durar muchas horas)
– Otros dispositivos médicos (por ejemplo, botella de oxígeno, pulsioxímetro…) o medicamentos recomendados por su médico.
B/ PROTOCOLO EMERGENCIA PARA HERMANOS DRAVET
Si tiene otros hijos, deberá hacer un plan para ellos por si usted tiene que ir al hospital. Considere tener un “padre de guardia” que acompañe al niño Dravet a urgencias. El otro padre o adulto designado puede quedarse con los otros niños para que puedan continuar con sus actividades.
Cuidado integral del paciente
La atención integral tiene como objetivo prevenir y/o tratar otros problemas que no sean las convulsiones y que vienen con el síndrome de Dravet: comorbilidades (presencia de uno o más trastornos además de la enfermedad primaria). El tratamiento puede no eliminar estos problemas, pero puede reducir su impacto en los pacientes y sus familias.
Aunque cada afectado presenta su propio cuadro clínico algunas condiciones secundarias son compartidas por casi todos los pacientes. La existencia de estas comorbilidades exige la actuación de otros profesionales en trabajo en equipo en el abordaje integral y multidisciplinar de la enfermedad.
A/ FIEBRE Y VACUNACIÓN
La fiebre es uno de los factores desencadenantes de ataques epilépticos más frecuentes. Por lo general, es bastante fácil controlar la fiebre con medicamentos. La fiebre tiene varias causas, pero la mayoría de las veces es un signo de infección.
La prevención de algunas infecciones con la vacunación puede ser útil.
1/ Infecciones y fiebre
Existe la sensación general de que los pacientes con síndrome de Dravet son más sensibles a las infecciones en comparación con otros. Sin embargo, no tienen ninguna deficiencia inmunológica conocida.
Se sabe que cada vez que una persona con síndrome de Dravet tiene fiebre, es más probable que experimente convulsiones. Si bien es cierto que algunas vacunas pueden causar una fiebre corta que puede desencadenar una convulsión, todavía se recomienda encarecidamente que los niños con síndrome de Dravet reciban todas las inmunizaciones infantiles de rutina.
Una dosis correcta de medicamentos antipiréticos (por ejemplo, paracetamol o ibuprofeno) por lo general es efectiva para reducir la fiebre. Recuerde pedirle a su médico de cabecera una receta de medicamentos antipiréticos.
Incluso si estos pacientes son sensibles a la fiebre, es imposible eliminar todas las enfermedades infecciosas. Deben tener una vida social lo más normal posible y no deben estar «aislados”.
Recientemente, una familia ha optado por ponerle un termómetro digital al paciente Dravet y monitorizar la temperatura corporal durante 24 horas. Esta familia ha sido capaz de anticipar las crisis epilépticas al inicio de una infección y han sacado unas conclusiones muy útiles a la hora de anticipar una convulsión generada por un cambio de temperatura corporal. Para más información sobre este estudio, pinche aquí.
2/ Vacunación
Los padres a menudo temen que la vacunación pueda desencadenar convulsiones, especialmente porque:
– Uno de los efectos secundarios de las vacunas es a veces causar fiebre
– La fiebre en algunos niños con epilepsia puede desencadenar una convulsión
– El primer episodio epiléptico para el síndrome de Dravet a menudo es desencadenado por una fiebre
Sin embargo, se recomienda encarecidamente vacunar a estos niños para protegerlos de enfermedades infecciosas graves.
Antes de la administración de la vacuna, hable sobre las pautas a seguir con su médico. En algunos casos conviene administrar con antelación un medicamento antipirético para la prevención de la fiebre o incluso administrar una dosis de algunos de los medicamentos antiepilépticos.
B/ FACTORES AMBIENTALES QUE PUEDEN PROVOCAR CRISIS EPILÉPTICA
Algunos pacientes con síndrome de Dravet son sensibles a diversos estímulos ambientales que pueden desencadenar convulsiones.
1/ Patrones Visuales
Los patrones visuales (repetición de formas visuales) pueden desencadenar convulsiones en algunos pacientes. Aunque no siempre es fácil evitarlos, existen algunas soluciones, como enmascarar completamente un ojo.
2/ Fotosensibilidad
Algunos pacientes con síndrome de Dravet pueden tener convulsiones provocadas por luces brillantes como luces estroboscópicas o el sol reflejado desde el agua en la playa. Para otros, los colores y las formas parpadeantes pueden ser importantes, como los videojuegos o los dibujos animados.
Consejos para estos pacientes:
– Evite las luces brillantes que parpadean
– Limite el tiempo que pasa frente a la televisión y otras pantallas electrónicas y reduzca su contraste. Siéntese lo más lejos posible de la pantalla y mantenga las luces encendidas en la habitación. Evita mirar pantallas en la oscuridad
– Gafas de sol regulares pueden ser útiles para el exterior. El valor de las gafas filtrantes especiales es incierto, pero puede ser útil para actividades al aire libre.
3/ Temperatura
El aumento de la temperatura corporal es un factor desencadenante de las convulsiones. Conviene evitar los cambios bruscos de temperatura corporal.
Puede ser conveniente evitar los deportes intensivos, el ambiente caluroso (estancias calurosas, automóvil…), los baños calientes o en la piscina. Algunos pacientes utilizan chalecos de enfriamiento o neoprenos para evitar los cambios bruscos en la temperatura corporal.
C/ TRASTORNOS ASOCIADOS
Todos los niños afectados por el síndrome de Dravet son únicos y puede experimentar uno o más de los siguientes trastornos asociados:
– Deterioro cognitivo
– Problemas de comportamiento y psicológicos
– Trastornos del sueño
– Problemas crecimiento y nutrición
– Perturbaciones de coordinación y ortopédicos
– Dificultades en la marcha
– Signos de disautonomía
– Problemas de salud dental
– Infecciones frecuentes en las vías respiratorias
– Problemas cardiovasculares
1/ Función cognitiva
Aunque la evolución del síndrome de Dravet es muy particular según cada paciente, las funciones cognitivas casi siempre se ven afectadas.
La dimensión, la gravedad y la naturaleza del deterioro cognitivo se pueden documentar mediante pruebas estandarizadas adaptadas a la edad. Esta prueba puede realizarse a intervalos a lo largo de la vida del paciente.
La presencia de alteraciones cognitivas importantes precisa la implicación de los profesionales de la psicopedagogía y/o neuropsicología para implantar terapias de estimulación y enriquecimiento ambiental.
2/ Problemas de comportamiento y psicológicos
Se pueden observar problemas de comportamiento y psicológicos en pacientes con síndrome de Dravet que pueden afectar su vida familiar y social.
Estos factores varían de un niño a otro y pueden presentar rasgos autistas, alteraciones graves de la conducta, comportamientos repetitivos, hiperactividad con o sin trastorno de la atención y problemas con la interacción social.
Se aconseja una vigilancia cercana al niño, especialmente cuando se asocia con hiperactividad, ya que pueden no percibir una situación como peligrosa.
Las actividades estereotípicas son aquellos movimientos y comportamientos repetitivos que pueden interferir con el juego estructurado.
La conducta desafiante puede hacer que los pacientes con Dravet sean difíciles de manejar en casa y en público. Algunas conductas desafiantes pueden estar vinculadas a una mala comunicación basada en el deterioro del lenguaje. Muchos pacientes con síndrome de Dravet tienen dificultades para interactuar adecuadamente con niños de su misma edad.
Hay pacientes que puede precisar de la toma de medicamentos específicos para tratar las alteraciones conductuales.
3/ Trastorno del sueño
Los pacientes Dravet pueden experimentar dificultades para dormir. Los problemas pueden incluir somnolencia excesiva o dificultad para conciliar el sueño, insomnio, apneas, terrores nocturnos o despertar prematuro. Existen muchas causas que incluyen convulsiones no reconocidas o efectos secundarios de algunos de los medicamentos antiepilépticos que pueden mejorarse mediante el ajuste de la dosis.
4/ Problemas de crecimiento y nutrición
Estos pueden incluir retraso del crecimiento, osteopenia (densidad ósea deficiente), escoliosis (curvatura de la espina dorsal), problemas con la alimentación, el apetito y la absorción de nutrientes, o dificultades con el proceso de la pubertad.
La mayoría de los trastornos de la alimentación, como la anorexia o la pérdida de apetito, son consecuencia de los tratamientos con medicamentos antiepilépticos. Los ajustes en la dosis pueden no ser fáciles debido al riesgo de más convulsiones. A veces un psicólogo puede ser de ayuda.
5/ Perturbaciones de coordinación y ortopédicos Dificultades en la marcha
Los pacientes con síndrome de Dravet a menudo están sujetos a alteraciones motoras y cambios posturales. Los síntomas pueden incluir trastornos en la marcha, escoliosis, deformidades del pie o de la rodilla.
Estos problemas pueden ser particularmente importantes durante la adolescencia. La marcha tiende a deteriorarse a partir de los nueve o diez años de edad, cuando los pacientes desarrollan gradualmente un patrón especial en cuclillas cuando caminan. Pueden tener dificultades para caminar largas distancias, aunque por lo general mantienen la capacidad de caminar por casa y en distancias cortas. Para los desplazamientos largos, puede ser necesario el uso de sillas o carros especiales.
Algunos pacientes Dravet han necesitado llevar unos DAFOS para iniciar la marcha a los 18 meses de edad. Consulte con su especialista.
Las habilidades motoras finas también se ven afectadas y pueden tener consecuencias en la lectura, la escritura y el dibujo.
6/ Signos de disautonomía
Alteraciones en la regulación de la temperatura, sudoración, ritmo cardíaco, la circulación sanguínea y digestión.
7/ Salud dental
Algunos pacientes suelen presentar problemas de salud dental, como el retraso en la erupción dental, bruxismo, maloclusiones, falta de piezas dentales, malformaciones, etc.
8/ Infecciones frecuentes en las vías respiratorias
Puede existir la posibilidad de infecciones frecuentes en las vías respiratorias, (oídos, nariz, senos paranasales, garganta, bronquios y pulmones.), del aparato digestivo o de las vías urinarias (estómago, intestinos, vejiga y riñones).
9/ Problemas cardiovasculares
Incluye frecuencia cardíaca rápida o lenta, ritmo cardíaco irregular y anormalidades en la estructura del corazón.
