Sin embargo, aun se desconoce por qué mutaciones similares en SCN1A causan fenotipos (síntomas) variados en los pacientes.
Para explicar esto, se barajan hipótesis no excluyentes como el efecto de la epigenética [modificaciones en la expresión de un gen causadas por alteraciones heredables que no son mutaciones, como por ejemplo un mayor grado de condensación de la cromatina (forma en la que se presenta el ADN en el núcleo celular; la cromatina condensada forma los cromosomas), que impida el acceso al gen de la maquinaria molecular encargada de su expresión] o la presencia de genes modificadores genes que afectan la expresión de otros genes). En este último caso, se han descrito ya algunos genes (ej. genes para receptores GABA o para canales de calcio, cloruro o potasio) con el potencial de modificar la expresión de SCN1A en ratones con síndrome de Dravet.
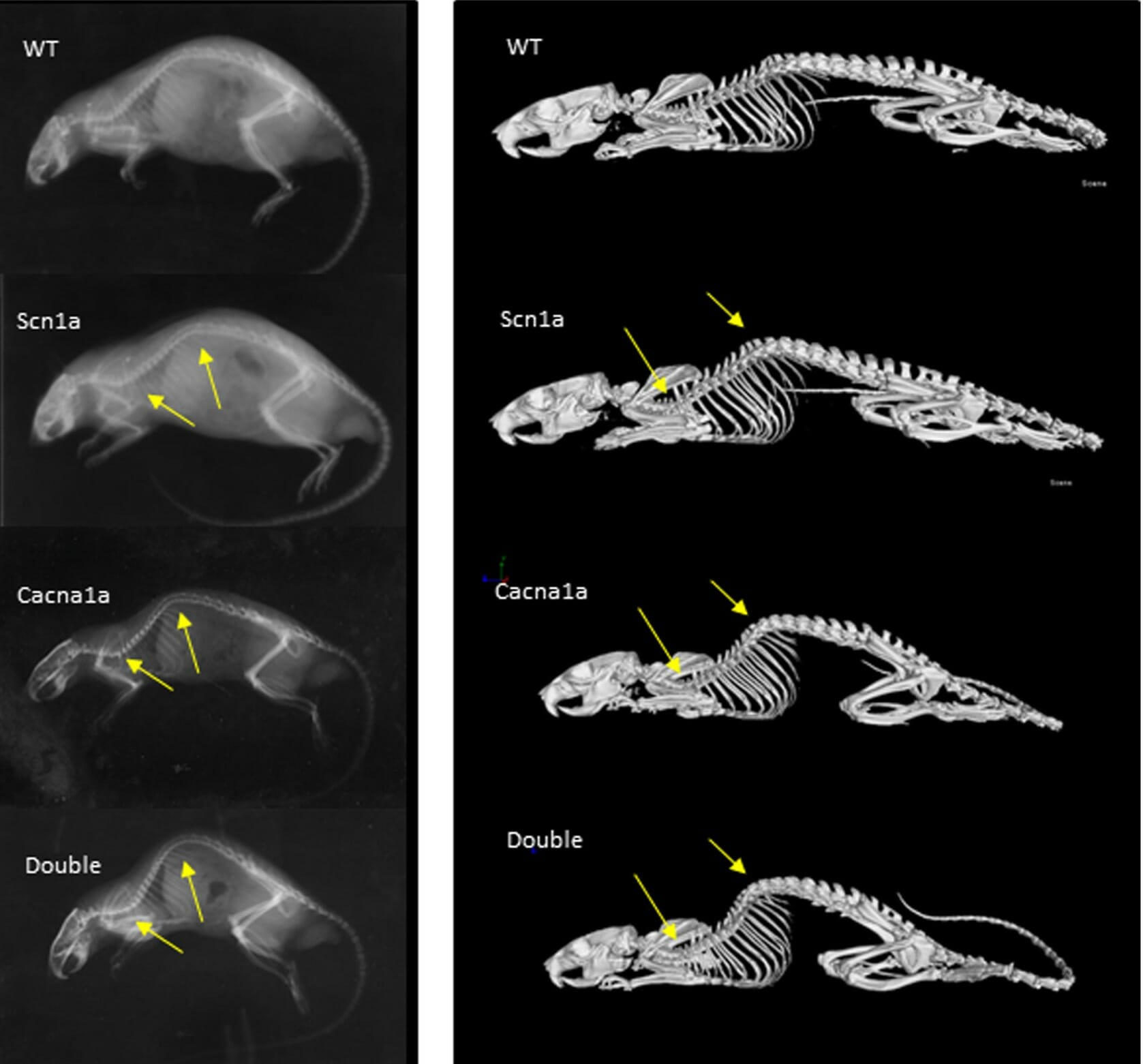
Un estudio reciente se ha centrado en analizar el efecto de la mutación de CACNA1A, un gen que codifica para el canal de calcio Cav2.1 y considerado como posible modificador de SCN1A, en ratas que presentan otra mutación en SCN1A. Ambos genes se expresan en las neuronas específicamente afectadas en pacientes con síndrome de Dravet, las interneuronas parvalbúmina.
Aquí os resumimos los resultados: 
 Las ratas con la doble mutación SCN1A/CACNA1A tenían un menor peso corporal que las ratas con mutación simple en SCN1A o las ratas sanas. Las ratas SCN1A/CACNA1A mostraron una curvatura espinal severa entre las vértebras cervical y torácica en comparación con la leve curvatura de las ratas SCN1A. Las ratas SCN1A/CACNA1A sufrieron crisis epilépticas tras un aumento de temperatura menor que las ratas SCN1A. Además, la duración de las crisis parecía mayor en las ratas SCN1A/CACNA1A. Sin embargo, los patrones de crisis tónico-clónicas generalizadas y electroencefalograma (EEG) fueron similares entre los dos grupos. Mientras que las ratas SCN1A no tenían crisis de ausencia, las ratas SCN1A/CACNA1A presentaron diferencias en los patrones ictales EEG de crisis de ausencia (el período ictal es la crisis propiamente dicha; el interictal es el período entre crisis; el postictal es el período inmediatamente posterior a una crisis en el que el cerebro se recupera). Las ratas SCN1A/CACNA1A sufrieron nuevos tipos de crisis espontáneas que no padecían las ratas SCN1A: crisis de ausencia, crisis de ausencia con componentes mioclónicos en párpados y extremidades, crisis tipo mioclónico, crisis clónicas espontáneas.
Las ratas con la doble mutación SCN1A/CACNA1A tenían un menor peso corporal que las ratas con mutación simple en SCN1A o las ratas sanas. Las ratas SCN1A/CACNA1A mostraron una curvatura espinal severa entre las vértebras cervical y torácica en comparación con la leve curvatura de las ratas SCN1A. Las ratas SCN1A/CACNA1A sufrieron crisis epilépticas tras un aumento de temperatura menor que las ratas SCN1A. Además, la duración de las crisis parecía mayor en las ratas SCN1A/CACNA1A. Sin embargo, los patrones de crisis tónico-clónicas generalizadas y electroencefalograma (EEG) fueron similares entre los dos grupos. Mientras que las ratas SCN1A no tenían crisis de ausencia, las ratas SCN1A/CACNA1A presentaron diferencias en los patrones ictales EEG de crisis de ausencia (el período ictal es la crisis propiamente dicha; el interictal es el período entre crisis; el postictal es el período inmediatamente posterior a una crisis en el que el cerebro se recupera). Las ratas SCN1A/CACNA1A sufrieron nuevos tipos de crisis espontáneas que no padecían las ratas SCN1A: crisis de ausencia, crisis de ausencia con componentes mioclónicos en párpados y extremidades, crisis tipo mioclónico, crisis clónicas espontáneas.
Este trabajo pone en evidencia la importancia de utilizar técnicas de secuenciación de nueva generación, con las que se pueden analizar grandes cantidades de material genético, o incluso el genoma completo, en el estudio de pacientes con enfermedades genéticas.
Aunque con limitaciones puesto que aun queda mucho por conocer acerca de los efectos de la epigenética y los genes modificadores en el Dravet, el estudio genético detallado puede ayudar a identificar las causas de los síntomas que padece un paciente. Un diagnóstico genético correcto y completo ayudará a mejorar la estrategia terapéutica del paciente, con el beneficio físico, emocional y económico que esto conlleva para el paciente, la familia y el sistema sanitario.
Podéis leer el estudio, en inglés, AQUÍ.
Un sueño, una meta.