
Manejo de la vacunación en un niño asintomático con una nueva variante en el gen SCN1A
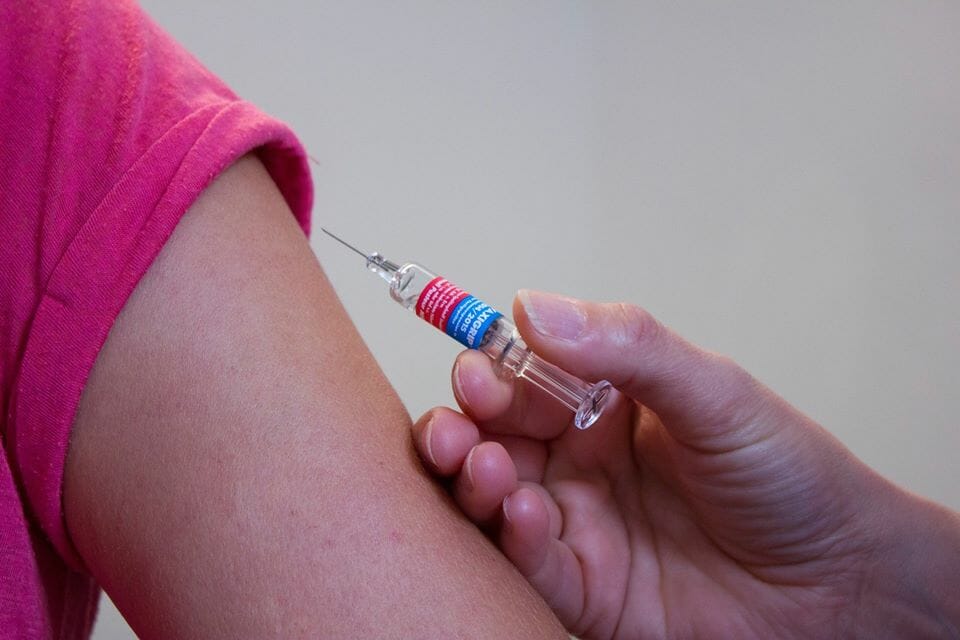
Os contamos un caso médico en el que el análisis genético familiar y la medicina personalizada han prevenido las crisis epilépticas en una paciente asintomática pero con una mutación en el SCN1A.
¡Muy interesante!
Aproximadamente el 85% de los niños con Dravet presentan una mutación en el gen SCN1A. El 95% de esas mutaciones son de novo, lo que significa que no son heredadas de los padres. Cuando la mutación es heredada, el padre o la madre que tiene la mutación suele presentar síntomas leves. Este es el caso de una familia australiana, y los médicos nos cuentan la historia en este artículo, el cual explicamos a continuación.
A los 12 meses de edad, un niño aparentemente sano tuvo su primera crisis epiléptica 27 horas después de administrársele las vacunas Hip (Haemophilus influenzae b), MenC (enfermedad meningocócica) y TV (triple vírica: sarampión-rubeola-parotiditis). Como sabéis, las vacunas pueden dar lugar a crisis epilépticas en pacientes Dravet. Esto no se debe a la vacuna en sí, sino a la posible fiebre a la que pueda dar lugar. En este caso, el niño ingresó en urgencias con 38.5ºC de fiebre. Su caso se complicó con un edema cerebral y bronconeumonía y, desafortunadamente, el niño murió.
Análisis genéticos del niño y de su madre* permitieron identificar en ambos una mutación en el gen SCN1A que nunca antes se había detectado: NM_001165963.1: c.2866A>G; p.(Met956Val). Esta mutación missense (de cambio de sentido) ha sido clasificada como posiblemente patógena por el Colegio Americano de Genética Médica, y podría dar lugar a la reducción de los niveles de Nav1.1, el canal de sodio al que da lugar el gen SCN1A, en la superficie neuronal, como ocurre con la mutación ya conocida en el mismo aminoácido de Nav1.1: Met956Thr.
Doce meses más tarde del fallecimiento del pequeño, la mujer tuvo otro bebé, en este caso una niña, la cual también presentó la misma mutación en SCN1A (Met956Val). Aunque la niña no presentó síntomas, los médicos decidieron tratarla de manera preventiva con valproato (20 mg/kg/día) para reducir el riesgo de epilepsia. Además, se le administró paracetamol y clobazam (0.3 mg/kg/día) tras sus vacunas de los 6, 12 y 18 meses. Actualmente, a sus 18 meses, la niña no ha presentado ninguna crisis epiléptica, y se planea terminar su medicación con valproato cuando cumpla los 2 años de edad.
Este caso recalca la importancia del análisis genético y la medicina personalizada en los pacientes con epilepsia y síndrome de Dravet. Quizá queráis compartir con vuestro médico este artículo porque, aunque el tratamiento de esta niña no tiene por qué ser válido para todos los pacientes, al menos este caso sirve como ejemplo de posibles procedimientos a seguir. Sabemos de al menos un caso entre nuestras familias en que el paciente tuvo 0 crisis tras seguir tratamiento con clobazam el día anterior, el mismo día y el día siguiente a la vacuna.
Un sueño, una meta.
.
*La madre tenía epilepsia cuando niña. Sus crisis fueron tratadas con valproato. Sus crisis desaparecieron 4 años antes de abandonar la medicación, lo cual se produjo 5 años antes de dar a luz a su primer hijo. Ella no presentó ningún problema de desarrollo.
Hubo también más casos de epilepsia en la familia: la hermana mayor y el padre de esta mujer.