
Hoy en día, el diagnóstico del síndrome de Dravet se basa en el consenso conseguido entre los expertos. Sin embargo, la profesora Ingrid Scheffer y su equipo buscan definir un diagnóstico basado en la evidencia de los datos procedentes de un gran grupo de pacientes.
Hoy la profesora Ingrid Scheffer, Amy Schneider y el Dr. Wenhui Li nos cuentan en primera persona de qué trata el nuevo artículo que han publicado con acceso abierto para todo el mundo gracias a una beca otorgada por la Fundación Síndrome de Dravet:
«La clasificación de los síndromes epilépticos se consigue a partir de la identificación de un grupo de pacientes que comparte un patrón similar de inicio de las crisis, los tipos de crisis, el perfil cognitivo y las comorbilidades asociadas como problemas de sueño, comportamiento, marcha y riesgo de mortalidad. La Dra. Charlotte Dravet identificó astutamente el síndrome que lleva su nombre en un pequeño grupo de pacientes en 1978.
En la actualidad hay miles de pacientes con síndrome de Dravet en todo el mundo y más del 90% presentan variantes patogénicas en el gen SCN1A. La necesidad no cubierta de un tratamiento que promete transformar el grave resultado a largo plazo de este síndrome significa que necesitamos desesperadamente un diagnóstico temprano y preciso.
Esto nos llevó a preguntarnos si un gran grupo de pacientes con síndrome de Dravet realmente tenía las características que enseñamos a los médicos y a las familias como típicas para el diagnóstico de este trastorno. El diagnóstico precoz es fundamental para que los pacientes no pierdan la oportunidad de recibir terapias específicas.
En nuestro estudio se analizó cuidadosamente la evolución temprana de 205 pacientes con síndrome de Dravet con mutaciones en el gen SCN1A. Descubrimos que mucho de lo que se enseña no es del todo correcto.
Algunos ejemplos de nuestros hallazgos son que la edad de inicio de las crisis epilépticas puede ser de hasta 20 meses; que en más de la mitad de los pacientes la primera crisis epiléptica es una crisis tónico-clónica en comparación con sólo un tercio que presenta una crisis hemiclónica; que sólo el 55% tiene fiebre con su crisis inicial y sólo un tercio presenta un estado epiléptico, donde la crisis dura más de 30 minutos. En siete infantes el tiempo transcurrido entre la primera y la segunda crisis fue de hasta 6 meses, caso en el que a menudo no se consideraría un diagnóstico de síndrome de Dravet (véase la figura).
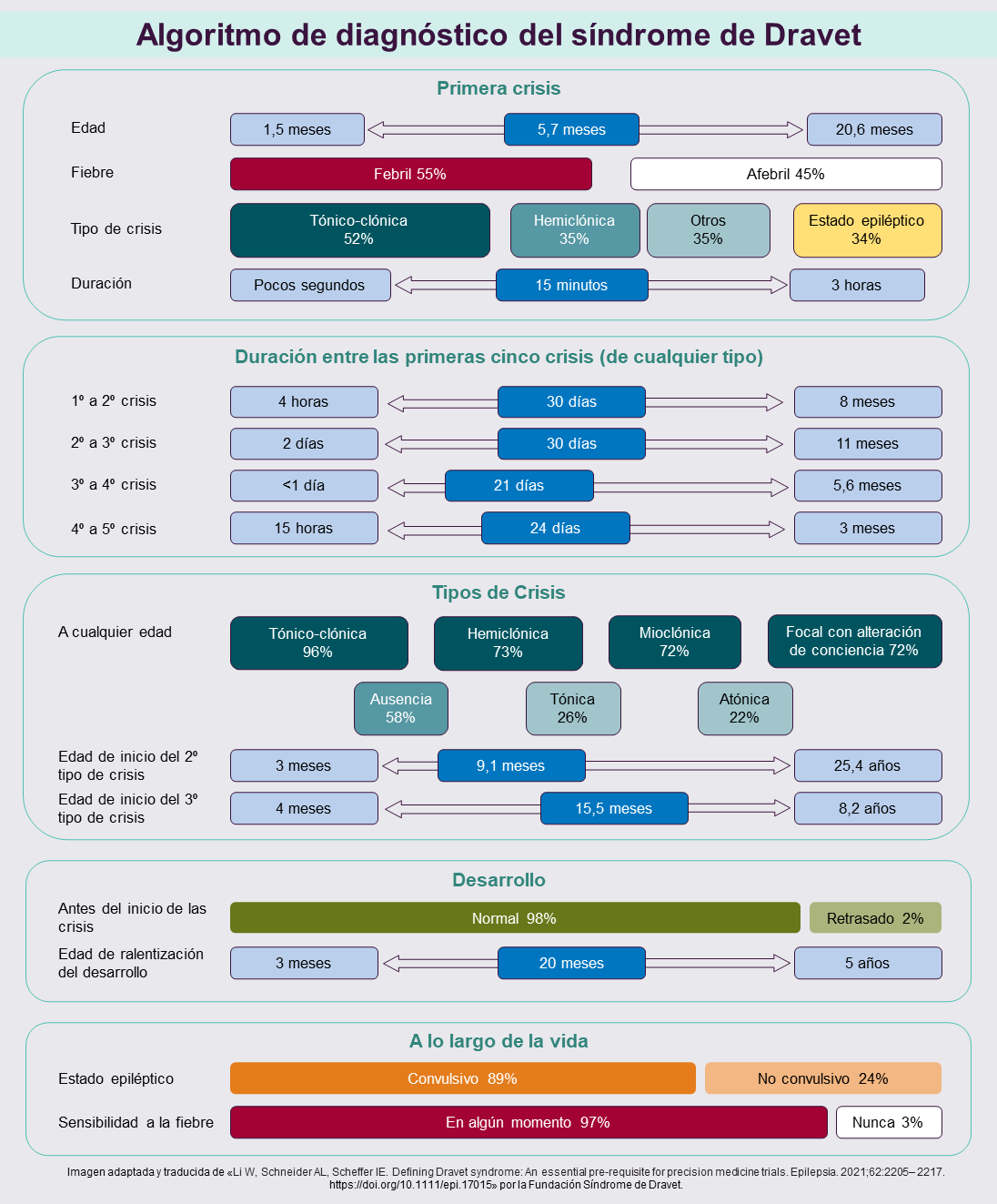
Este análisis de cómo se presenta la enfermedad allana el camino para una definición cuidadosa del síndrome basada en una gran cohorte de casos y hará que la definición del síndrome de epilepsia pase de la visión de consenso de los expertos a la definición de la enfermedad basada en la evidencia. Nuestro trabajo destaca los datos que necesitamos para informar sobre el diagnóstico precoz y permitir que se administren las terapias específicas adecuadas inmediatamente ―desde los medicamentos anticonvulsivos correctos actualmente disponibles (y no los incorrectos) hasta la promesa de las próximas terapias génicas».
Este artículo ha sido escrito por Ingrid E. Scheffer para la Fundación Síndrome de Dravet y traducido y adaptado por la Fundación Síndrome de Dravet.
Pincha AQUÍ para acceder al artículo científico original en inglés, publicado en acceso abierto en la prestigiosa revista Epilepsia gracias a una beca de la Fundación Síndrome de Dravet otorgada al equipo de la Prof. Scheffer.
Os recordamos que la CONVOCATORIA para acceder al Programa de Apoyo a las Publicaciones Científicas en Acceso Abierto de la Fundación Síndrome de Dravet («FSD Open Access Publication Support Program») sigue ABIERTA DURANTE TODO EL AÑO 2021.
¿Quieres ser tú el próximo en publicar tu investigación en síndrome de Dravet?
¡Solicita tu beca pinchando AQUÍ!




