
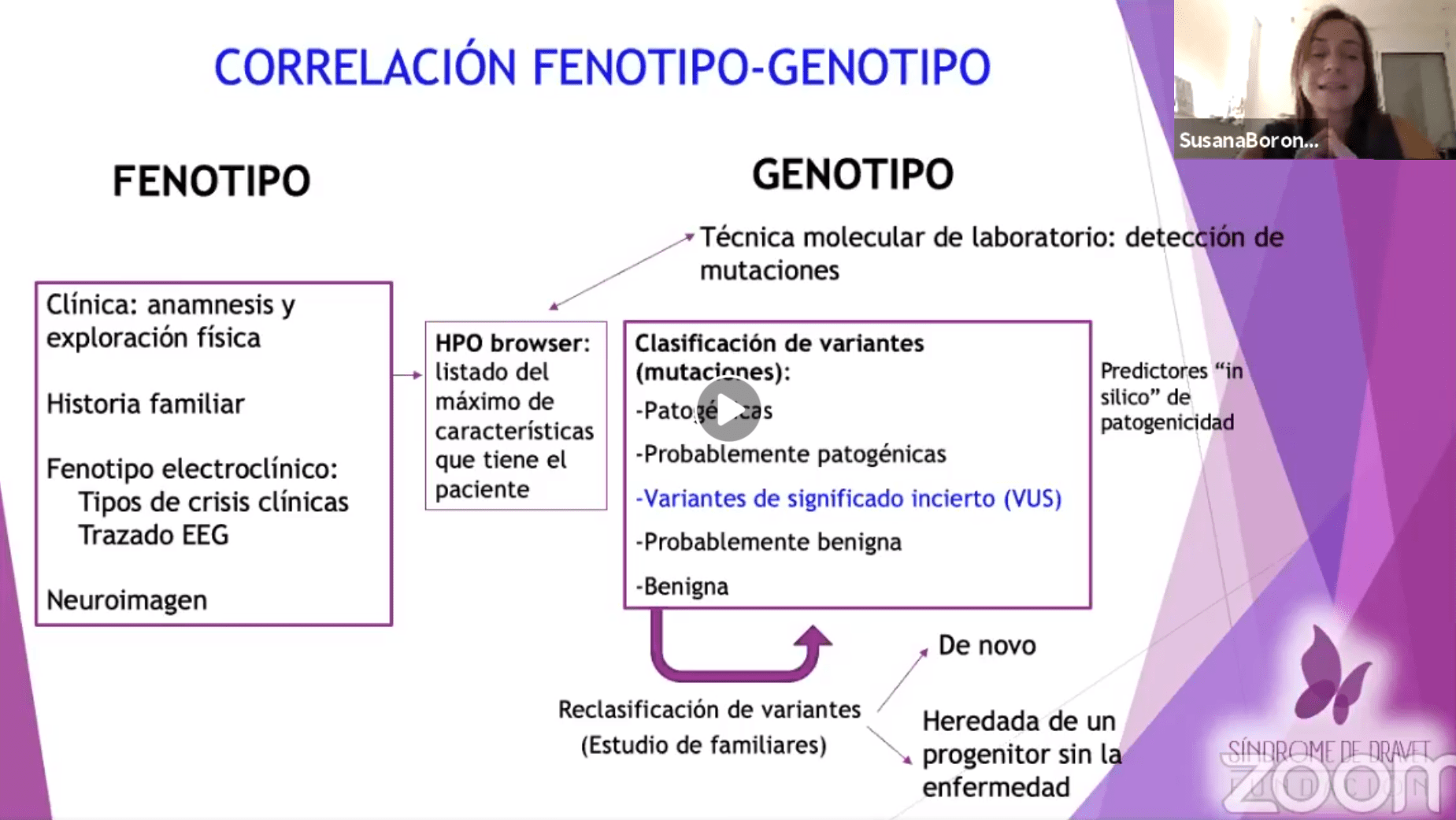

Webinar Gestión de Comportamientos Desafiantes en el Síndrome de Dravet
🧠✨ Con un enfoque personalizado, Lorena Rodríguez de Neurokid nos enseña en este webinar a entender y manejar ...

La Fundación Síndrome de Dravet celebra la inclusión en el debate político de la jubilación anticipada para cuidadores de grandes dependientes que trabajan
Madrid, 24 de abril de 2024.- La Fundación Síndrome de Dravet celebra la inclusión en el debate político, ...

Un estudio reciente explora las dificultades conductuales en el síndrome de Dravet
Un nuevo estudio resalta la necesidad de un enfoque integral y personalizado para el manejo de las dificultades ...

Webinar Fisioterapia en el síndrome de Dravet Una guía para padres y cuidadores
Acompaña a Fernando Piñango en un viaje por la fisioterapia y descubre técnicas y consejos prácticos en este ...

Unidos por la esperanza en la Reunión Anual de Familias 2024
Nuestra Reunión Anual de Familias 2024 fue un éxito rotundo en Madrid, con familias, profesionales y ponentes compartiendo ...

European Dravet Syndrome Conference 2024: Avances y Esperanza
Descubre los últimos avances en la lucha contra el síndrome de Dravet en nuestro resumen de la European ...

Webinar: Día Mundial de las Enfermedades Raras
Comparte este webinar con los más peques para que puedan aprender sobre el síndrome de Dravet y otras ...

Un nuevo vector de terapia génica para el síndrome de Dravet muestra resultados prometedores en ratones
Un nuevo vector de terapia génica demuestra ser eficaz en un modelo de ratón Dravet, aumentando la supervivencia ...

Los oligonucleótidos antisentido (ASO) y su papel en el tratamiento del síndrome de Dravet
¿Sabías que los oligonucleótidos antisentido podrían mejorar el síndrome de Dravet? Estas moléculas se unen al ARN mensajero ...

¿Cómo afecta el síndrome de Dravet al lenguaje y la comunicación?
Un nuevo estudio revela el impacto devastador del síndrome de Dravet en el lenguaje y la comunicación.






